{kind=link}
{kind=link}
{kind=link}
Syndrome Description
Learn more about M-CM
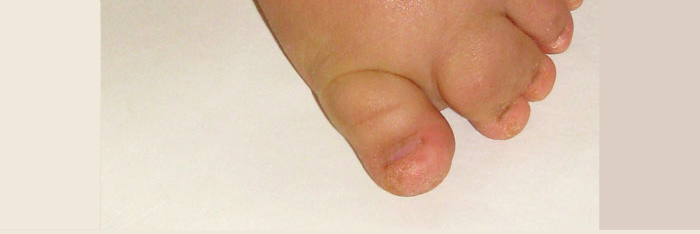
Most of the conditions commonly misdiagnosed in individuals with M-CM include those with overgrowth and vascular malformations seen in the skin.
PROS stands for PIK3CA-Related Overgrowth Spectrum. This descriptor encompasses a variety of conditions caused by mosaic PIK3CA mutations, including M-CM. Some patients with mosaic PIK3CA mutations will fall clearly within clinical diagnostic criteria for a particular named syndrome, and others will have characteristics that overlap with these syndromes. The PROS conditions that M-CM patients most commonly overlap with are CLOVES and Klippel-Trenaunay Syndrome.
CLOVES syndrome is named by an acronym which stands for: C (congenital) L (lipomatous) O (overgrowth) V (vascular malformations) E (epidermal nevi) S (scoliosis and other skeletal involvement). This condition was first described in 2007 in a group of individuals who had previously been diagnosed with Proteus syndrome. CLOVE(S) can cause capillary malformations, lipomas and hemihypertrophy although it is not typically associated with significant neurological findings. CLOVES is caused by a mosaic mutation in PIK3CA. This is the same gene that is mutated in M-CM.
Klippel-Trenaunay syndrome (KTS) is characterized by a triad of findings including slow-flow vascular malformations involving the veins and capillaries, an abnormally developed lymphatic system, and hemihyperplasia. Typically, the limbs and/or trunk are affected in an isolated manner. Overgrowth of an affected body part is present at birth and is often severe. Port wine stains and varicose veins are common. Of all the diagnoses listed here, KTS is probably the one that individuals with M-CM are misdiagnosed with the most. Generally, individuals with KTS do not have macrocephaly and do not have significant developmental delay or neurologic problems. Some Klippel-Trenaunay patients have been found to have mosaic PIK3CA mutations.
Parkes-Weber syndrome (PWS) is similar to Klippel-Trenaunay syndrome. However in PWS, the vascular anomalies are fast-flow lesions involving abnormal connections between the arteries and veins (arterial-venous malformations or AVMs). Some patients with PWS have been found to have mutations in a gene called RASA1.
Sturge-Weber syndrome (SWS) is characterized by capillary malformations of the meninges which is usually accompanied by characteristic facial port wine stains (mostly on the upper eyelid and forehead) which exist in a specific distribution pattern related to the facial nerve. The disorder often has neurological problems or seizures due to the involvement with the brain. There may occasionally be capillary malformations of the skin below the head and neck. SWS is caused by mosaic mutations in a gene called GNAQ.
Cutis Marmorata Telangiectatica Congenita (CMTC) is characterized by generalized or segmental marbling of the skin due to prominent blood vessels.This looks like cutis marmorata but is much more pronounced. Other vascular malformations such as hemangiomas, salmon patches and varicose veins can also occur in patients with CMTC. The cause is unknown and while it is often an isolated finding, there are risks of other associated anomalies including glaucoma and limb asymmetry (most often hemihypoplasia, or undergrowth of the affected limb). In general, isolated CMTC looks very different than M-CM. Because of this, it is not clear if the disorders share anything more than a superficial resemblance. Neurological complications such as macrocephaly, cerebellar overgrowth, hydrocephalus and developmental delay are less common compared to M-CM.
Beckwith-Wiedemann syndrome (BWS) is a relatively common overgrowth disorder that includes the following as frequent features: large mouth and tongue, omphalocele, umbilical hernia, ear creases or pits, newborn hypoglycemia, capillary malformations and hemihyperplasia. Genetic testing can identify most cases. Individuals with BWS do not typically have severe neurological complications unless they experienced severe hypoglycemia in the newborn period. BWS does not tend to cause the widespread vascular abnormalities of the skin seen in M-CM. Likewise, patients with M-CM generally do not have an increased risk of omphalocele.
Proteus syndrome (PS) is a rare and complex disorder that involves overgrowth of body parts along with abnormalities of the blood vessels and lymphatic system, localized areas of bone overgrowth (hyperostoses), epidermal nevi, connective tissue nevi and other connective tissue components such as the muscles and fat. Patients may have benign, fatty tumors (lipomas) or lack of fat under the skin in certain parts of the body. This is a rare, progressive condition where children are usually born without any obvious deformities and acquire them over time. This distinguishes PS from many other overgrowth syndromes, including M-CM.
Formal diagnostic criteria for PS exist. However, because the disease is poorly understood and rare, many clinicians, even experienced geneticists, may struggle with making the diagnosis accurately. In 2011, a mutation in the AKT1 gene was associated with PS. This mutation was seen in some but not all of the tissues of affected individuals, confirming that this condition is due to genetic mosaicism. It has been suggested in the past that some individuals with PS have mutations in a gene called PTEN. However, those who clearly have PS when strict use of the diagnostic criteria is applied do not have an identifiable defect in the PTEN gene. This recent discovery clarifies the molecular differences between PS due to the AKT1 gene and Proteus-like syndromes due to mutations in PTEN.
This new discovery of the association of the AKT1 gene and PS should further dispel any potential direct link between PTEN mutations and PS.
Hemihyperplasia-Multiple Lipomatosis syndrome (HHML) was first described in 1998 in a group of individuals with moderate body asymmetry and lipoma-like lesions under the skin. These individuals did not meet the clinical diagnostic criteria for Proteus syndrome despite many overlapping features.
PTEN-Related Hamartoma Syndromes
PTEN is a gene that, when altered, can cause different disorders that are characterized by the development of noncancerous tumor-like growths called hamartomas. Together, the disorders caused by PTEN are called PTEN-related hamartoma syndromes and are summarized below. Some but not all cases of the following conditions are due to identifiable changes in the PTEN gene.
MPPH syndrome (megalencephaly-polymicrogyria-polydactyly-hydrocephalus) is a condition that was first described in 2004. MPPH shares some brain characteristics M-CM including megalencephaly, polymicrogyria and hydrocephalus, as the name implies. It lacks vascular anomalies and hemihyperplasia. MPPH is caused by mutations in AKT3, CCND2, or PIK3R2. The gene discovery for MPPH was published in the same paper as that for M-CM.
Costello syndrome is characterized by delayed development and intellectual disability, distinctive facial features, curly or sparse fine hair, loose folds of extra skin (especially on the hands and feet), hypotonia and unusually flexible joints. Heart abnormalities are common including tachycardia, structural heart defects, and hypertrophic cardiomyopathy. Infants with Costello syndrome tend to be larger than average at birth including macrocephaly with progressive frontal bossing. They do not have the same vascular skin changes seen in M-CM nor the characteristic malformations of the digits.
There is considerable overlap with M-CM in Costello syndrome in terms of neurological findings on brain imaging studies. Common neurological findings in Costello include Chiari I malformations, progressive and evolving ventriculomegaly/hydrocephalus, posterior fossa crowding and cerebellar tonsillar herniation. Costello syndrome is caused by mutations in the HRAS gene and clinical testing is available. The disorder is different enough from M-CM that an experienced clinical geneticist should be able to differentiate the conditions.
Sotos syndrome is an overgrowth condition with features including macrocephaly, characteristic facial features, overgrowth of the body, learning disabilities, behavioral problems, seizures, scoliosis, congenital heart and kidney defects. Asymmetry or hemihyperplasia, syndactyly/polydactyly, and vascular skin abnormalities are not common features in this syndrome. Sotos syndrome is due to defects in the NSD1 gene and genetic testing is available.
Simpson-Golabi-Behmel syndrome (SGBS) is a genetic condition associated with macrosomia, distinctive facial features, macrocephaly, heart defects, gastrointestinal and genitourinary abnormalities, musculoskeletal findings including congenital hip dislocation, and digital anomalies including polydactyly and syndactyly. Affected individuals have variable neurologic involvement, ranging from normal to severe mental retardation with or without structural brain anomalies. SGBS is due to changes in the GPC3 gene and genetic testing is available. Unlike the other disorders on this list, SGBS mostly affects boys due to the fact that it is an X-linked condition. This means that the GPC3 gene is located on the X chromosome.

Order brochures or download a PDF
Guidelines from published research literature
Guidance for clinical genetic testing

Explore the results of a patient survey conducted in 2012

Explore the research literature related to M-CM

An extensive list of resources